
|
Recherche de similaritķs dans les banques
|
 |

|
|
|
|
|
|
|
INTRODUCTION

|
Après avoir déterminé la séquence d'une portion d'ADN, l'une
des premières questions est « Quelqu'un a t-il déjà
rencontré ce type de séquences ? ». Pour y répondre,
il faut aller faire des recherches dans les banques de séquences
nucléiques ou protéiques.
Pour cela de nombreux programmes de recherche de séquences similaires
à une séquence d'intérêt sont disponibles mais
il faut noter que ces programmes:
|
|
|
 |
la plupart utilisent des heuristiques pour rendre la recherche rapide :
ceci à pour conséquences une perte de la rigueur de la comparaison
(des programmes plus rigoureux existent mais ils sont beaucoup plus lents).
|
|
|
|
|
peuvent « rater» une similarité importante (faux négatifs).
|
|
|
|
|
peuvent ramener des séquences dont la similarité avec la
séquence d'intérêt n'est pas significative (faux positifs).
|
|
|
|
|
constituent une aide pour identifier un échantillon de séquences
nécessitant une analyse plus poussée mais ne constituent
pas l'analyse.
|
|
|
|
|
L'ADN est constitué de 4 lettres (contre 20 pour les protéines):
la probabilité de « matcher » est donc beaucoup plus
importante lors de comparaisons ADN / ADN.
|
|
|
|
|
La comparaison de deux nucléotides répond le plus souvent
à une loi oui / non tandis que pour deux acides aminés, cette
comparaison est plus fine car elle peut être basée sur des
critères physico-chimiques, la similarité des codons d'ADN
ou sur des taux de mutations naturels.
|
|
|
|
|
Les banques protéiques sont beaucoup plus petites que les banques
nucléiques.
|
|
|
|
|
 |
|
|
Homologie vs Similarité
|
|
Lorsqu'on parle de recherche dans une banque de données on se réfère
souvent à une recherche d'homologie. Or le terme homologie
implique une notion d'évolution commune entre les deux partenaires
: ce n'est pas parce que deux séquences ont des nucléotides
ou des acides aminés identiques qu'elles ont forcément un
ancêtre commun. Il faut donc parler de recherche de similarité,
même si un fort taux de similarité (25% d'identité sur 100
acides aminés) est considéré comme une preuve d'une
homologie donc de l'existence d'un ancêtre commun.
|
|
|
|
|
|
Similarité globale vs similarité locale
|
|
Les premiers outils développés par Needelman & Wunch (J.
Mol Biol. 48:444-453, 1970) et Sellers (SIAM. 26:787-793, 1974) calculaient
un score de similarité globale, c'est à dire sur la
totalité de la séquence, entre les deux séquences
à comparer.
Ces algorithmes ne sont pas, en général assez sensibles pour
comparer des séquences très divergentes. La méthode
retenue par les programmes de recherche de similarité est de se
baser sur de courtes régions pour calculer une similarité
locale. Cette méthode à l'avantage d'être beaucoup
plus rapide.
Il existe trois programmes très répandus utilisant
les algorithmes de calcul de similarité locale :
|
|
|
|
Smith-Waterman
(J Mol Biol 147:195-197, 1981)
|
|
|
|
|
BLAST
(Altschul et al, J Mol Biol 215:403-410, 1990)
|
|
|
|
|
FASTA
(Pearson and Lipman, Proc Natl Acad Sci USA 85:2444-2448, 1988).
|
|
|
L'algorithme de Smith - Waterman permet une approche par programmation dynamique et
n'utilise pas d'heuristique. Il n'est pas utilisé en routine car
s'il est beaucoup plus sensible que BLAST ou FASTA, il est aussi 100 fois
plus lent.
FASTA est peut être plus sensible que BLAST pour des recherches dans les
banques nucléiques mais dans la plupart des cas, les deux programmes
peuvent être utilisés afin de faire la recherche la plus complète
possible. Il faut néamoins savoir que la sensibilité de
FASTA a un prix : une recherche via ce programme est beaucoup plus longue
et consomme plus de ressources.
|
|
|
|
|
|
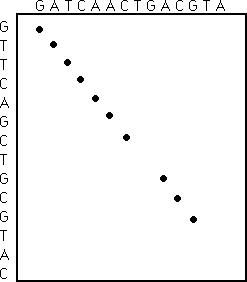
Dotplots
|
Les dotplots sont utilisés
- pour comparer visuellement deux séquences et détecter
les régions ayant une forte similarité.
- par les programmes de recherche de similarité
dans les premières étapes de recherche.
Dans un dotplot (figure 1), les deux séquences sont placées
le long des axes d'un graphique. L'intersection de chaque ligne et colonne
est marquée d'un point si la lettre est la même dans les deux
séquences.

Figure1 : dotplot simple
Une suite de points sur la diagonale indique les régions de similarité
entre les deux séquences. Si un oeil entraîné peut
distinguer les régions de similarité sur cette figure, il
est préférable d'appliquer des méthodes statistiques
qui permettent de mettre en évidence ces régions en éliminant
le bruit de fond, comme par exemple, utiliser un filtre qui autorise un
point uniquement si plusieurs bases successives « matchent ».
Dans la figure 2, on a représenté le même dotplot avec un
filtre qui ne place un point que si dans une fenêtre de 4 bases,
3 de ces 4 bases « matchent ».
Pour détecter des similarités plus lointaines, il peut être
utile d'utiliser une fenêtre plus grande (20 ou 30 et même
50 bases) et un pourcentage d'identités plus faible (par exemple
50%).
| G |
A |
T |
C |
A |
A |
C |
T |
G |
A |
C |
G |
T |
A |
| G |
T |
T |
C |
A |
G |
C |
T |
G |
C |
G |
T |
A |
C |
Figure 2 : Dot plot avec 75 %d'identité dans
une fenêtre de 4 bases
De toutes façons, avec des séquences réelles, les motifs ne
sont pas aussi évidents!
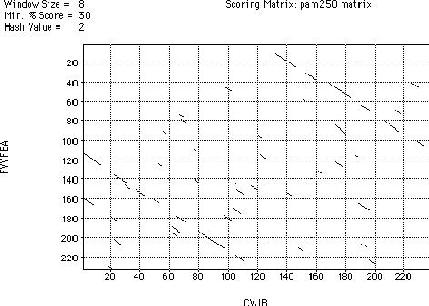
Figure3 : un dotplot avec 2 séquences de
230 AA
|
|
|
|
|
|

